固体系のエネルギー・バンド構造を量子コンピュータを用いて計算する手法に関する論文(プレプリント)を公開しました。
株式会社QunaSysの中川と、理化学研究所の吉岡信行特別研究員(元QunaSysインターン)、JSR株式会社の大西裕也マテリアルズ・インフォマティクス推進室次長、大阪大学の水上渉特任准教授(QunaSys技術顧問)は、変分量子アルゴリズムを用いた固体系のバンド構造計算に関する論文(プレプリント)を発表しました。
この成果は、QunaSysとJSR株式会社との共同研究 に基づくものです。
"Variational Quantum Simulation for Periodic Materials"
https://arxiv.org/abs/2008.09492
追記:2022年1月26日付で、Physical Review Research誌に掲載されました。
https://journals.aps.org/prresearch/abstract/10.1103/PhysRevResearch.4.013052
背景
物質の三態の一つである固体では、原子や分子が周期的に並び、その種類や並び方(結晶構造)によって様々な性質(物性)が現れます。物質の結晶構造からその物性を予測する固体物理学は新規材料・素材の開発において非常に重要であり、大学・産業界の双方で精力的な研究が行われています。現在、固体物理学の理論計算では密度汎関数法(DFT)が広く用いられており、数多くの成功を収めています。しかし、半導体のバンドギャップなどDFTが苦手とする計算領域が依然として存在することも知られており、DFTとは異なる「量子化学計算」の手法を固体物理学に適用する研究が注目され始めています。量子化学計算は、DFTと比べて (1) 経験的パラメータを含まない第一原理的な計算を実行できる (2) 手法を変えることで計算精度を系統的に改善することができる、といった利点を有しており、従来のDFTでは予測が難しかった物質や物性値の計算での活用が期待されています。
問題点
しかし、固体物質に量子化学計算の手法を適用した場合、必要な計算資源が非常に大きくなってしまうという問題があります。そこで、近年急速に開発が進んでいて、量子化学計算への応用が期待されているNoisy Intermidiate-Scale Quantum (NISQ)デバイスという量子コンピュータを活用できないかというアイディアが考えられます。NISQデバイスによる量子化学計算の研究ではこれまで分子系への適用のみが注目されており、固体物質への応用に関する知見は得られていませんでした。
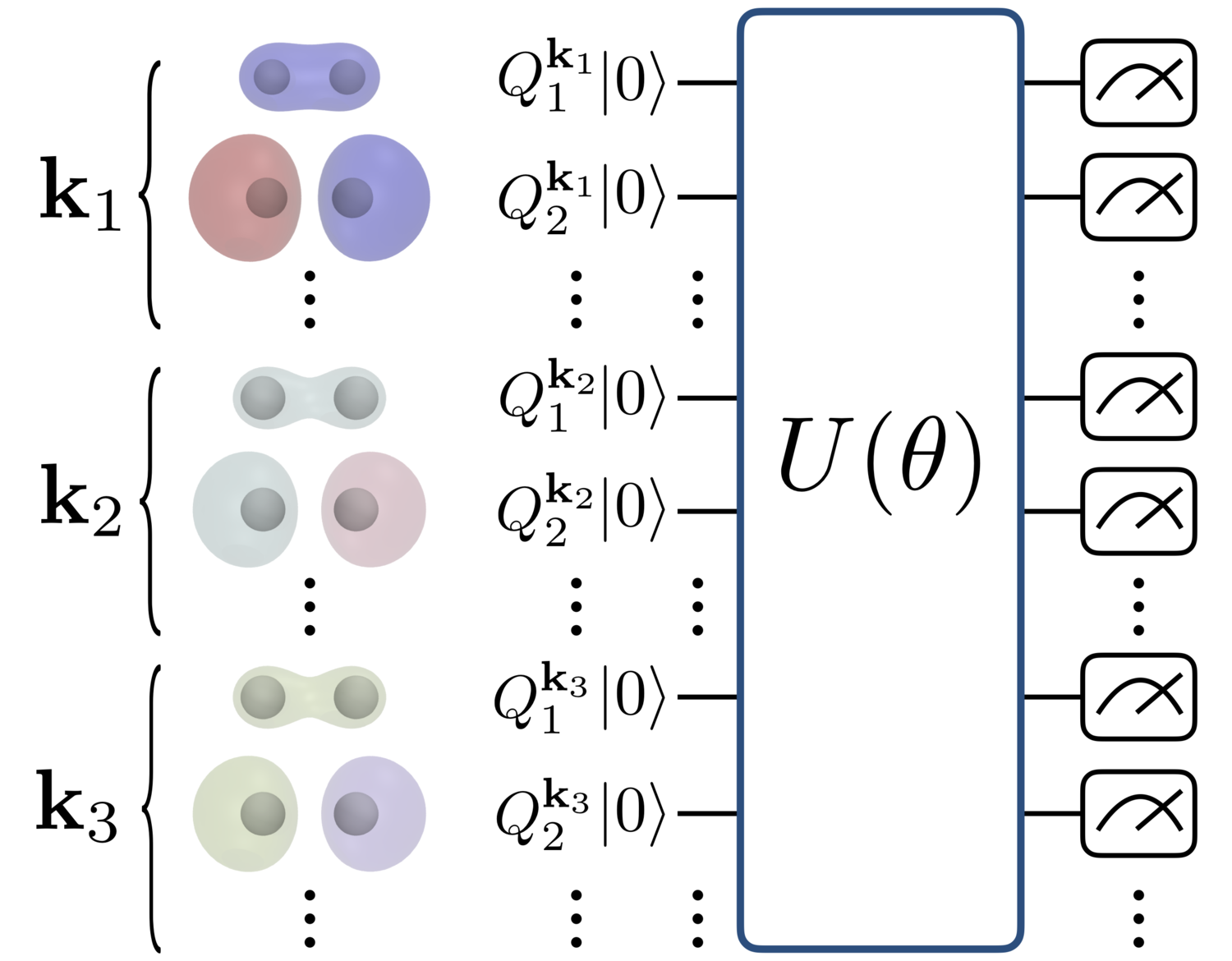
方法
本研究では、NISQデバイスを用いた量子化学計算アルゴリズムの代表例である変分量子固有値ソルバー(Variational Quantum Eigensolver; VQE)を用いて、水素原子が周期的に並んだモデル物質のエネルギーを計算するシミュレーションを行いました。具体的には、UCC-SD (unitary coupled-cluster singles and doubles)と呼ばれる試行波動関数を固体物質に拡張したものを用い、量子回路の出力を数値的にシミュレーションすることで、実際に量子コンピュータ上でVQEを実行した場合にどのようなエネルギー値が得られるのかを推測しました。
数値計算には、化学計算のオープンソースライブラリPySCF( https://github.com/pyscf/pyscf)とQunaSysの管理する量子回路の高速シミュレータQulacs(http://docs.qulacs.org/ja/latest/#)を用いました。
結果
数値シミュレーションの結果、VQEによって求められたモデル物質の基底状態(最も安定な状態)のエネルギーは、別の独立な手法で求めた厳密な値とよく一致する傾向がみられました。さらに、水素原子間の距離を変えていくと、古典コンピュータを使った量子化学計算の代表的な手法であるCCSD法が誤ったエネルギーを返すような領域でも、VQEでは厳密な値に近いエネルギーを求められることがわかりました。さらに、基底状態に加えて、量子部分空間展開法を応用することで(擬)バンド構造の計算を実現しました。その結果、標準的な第一原理計算法であるDFT(PBE)よりもエネルギーギャップを正確に見積もれることを確認しました。
展望
本研究(プレプリント)により、量子コンピュータを用いた固体物質の量子化学計算へ向けた第一歩となる知見が得られました。量子コンピュータの応用先が分子系だけではなく固体系へとさらに広がり、より幅広い産業分野へのインパクトが期待されます。