Qamuy / 量子化学計算クラウドサービス
Qamuyは量子アルゴリズムを用いて量子化学計算を行うクラウドサービスです。
量子アルゴリズムの専門知識は不要です。
Want to know more?

Qamuyの用途
量子コンピュータで量子化学計算を実行
電池材料や光応答材料などユーザーが興味ある材料で、量子コンピュータ上での量子化学計算が可能です。
現時点ではハードウェアの能力に制約がありますが、将来的に量子コンピュータをどう活用できるか、量子化学計算を通じて検証することができます。
量子コンピュータのパワーを評価
Qamuyで実際に量子化学計算を行うことで、必要な計算リソースを見積可能です。
実機での計算時間の見積結果の活用で、近い将来のいつ頃からどんな用途で量子コンピュータが活用可能になるか、エビデンスを持って推測可能です。
カスタムの量子
ワークフロー構築
Qamuyの豊富な量子化学計算ライブラリを利用できるだけでなく、各企業独自のコンポーネント組み込みやカスタムワークフローを構築できます。Qamuyがお客様に最適化したツールへと進化し、来たる量子コンピュータの時代にパワーを最大限活用できるようになります。
Qamuyの利用フローイメージ
Qamuyは、量子化学計算のインプットを量子回路に翻訳し、シミュレータや実機上での計算をシームレスに行うことができる量子化学計算クラウドです。
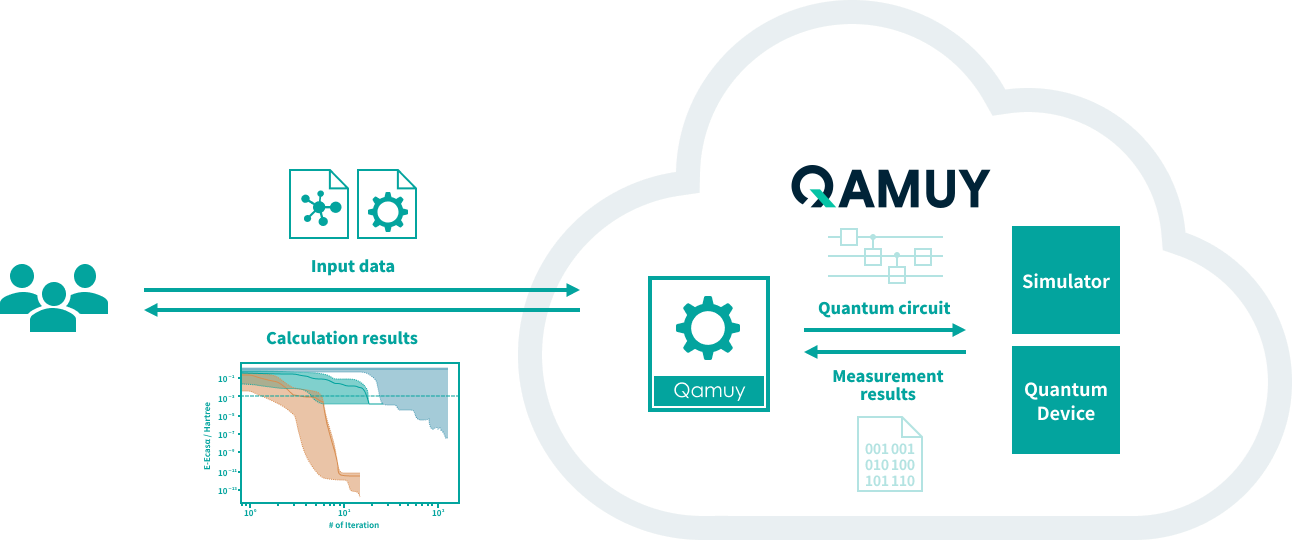
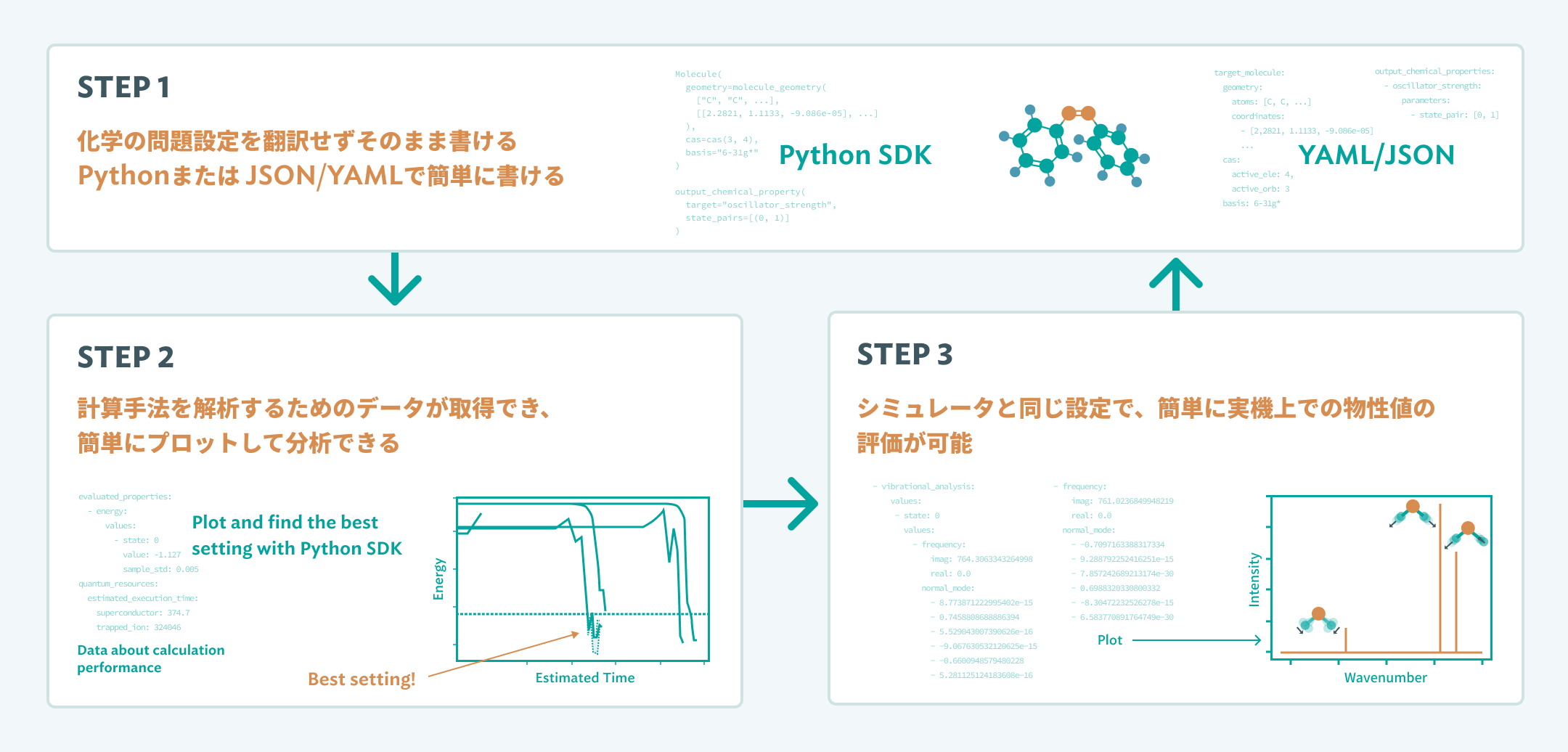
STEP 1計算したい課題を準備
量子回路の専門知識は必要ありません。
化学計算に用いるインプットをQamuyが量子回路に翻訳します。
STEP 2シミュレータ上で量子
計算手法を最適化
量子コンピュータ上で量子化学計算を行う場合、最適化が必要なパラメータが多数あります。Qamuyを使うことで最適化を直感的に行うことができ、その過程を通じて計算手法への理解が深まります。
STEP 3実機上で計算を実行
計算手法の最適化が完了したら、いよいよ実機での計算を行うことができます。Qamuyには、実機での計算をサポートする機能が揃っています。
Qamuyの機能
Qamuyには、産業上重要な量子化学計算を行うための最先端のアルゴリズムが多数実装されています。
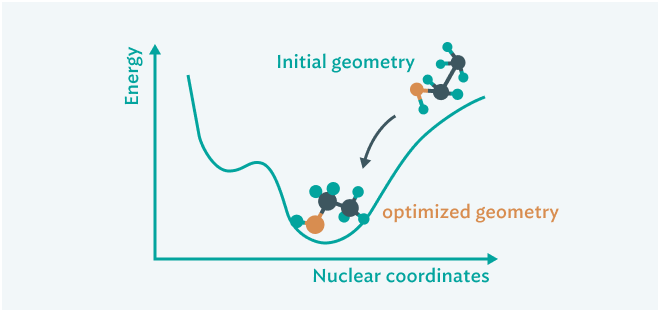
構造最適化
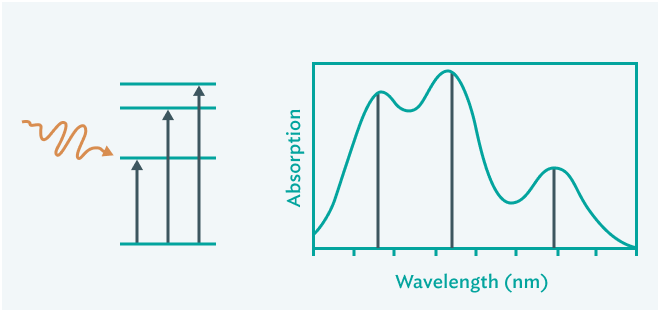
光吸収スペクトル
分子動力学
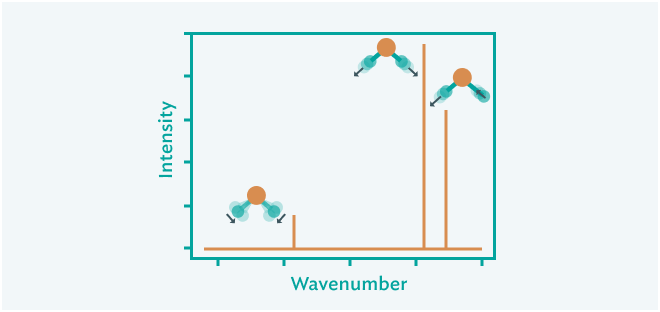
振動数解析
非断熱動力学
バンド構造
量子アルゴリズム
- 変分量子ソルバー(VQE法・SSVQE法・VQD法)
- 分子軌道最適化
- 試行波動関数(Hardware efficient/Symmetry preserving/UCCD/UCCSD/Fermionic adaptive)
- 量子回路パラメータ最適化
物理量の計算
- エネルギー
- エネルギー微分
- 双極子モーメント
- 振動子強度
- 非断熱結合
- 振動解析
- バンド構造
- 構造最適化
量子回路シミュレーション
- 状態ベクトルシミュレーション
- 厳密期待値計算
- 期待値のサンプリング評価
- ノイズ入りシミュレーション*
高度な計算最適化
- 量子ビット数削減
- 観測量グルーピング
- サンプリング戦略
実機利用・評価
- 量子コンピュータ実機上での計算*
- 実機での計算リソース見積もり
- 量子アルゴリズムの性能評価情報
分析・連携機能
- JSON/YAMLによるデータ入出力
- SDKを用いた自動化・効率化
- 結果データの可視化
- 外部ツールとのデータ連携
USE CASES / 01
光化学反応計算への応用検討
CLIENT
三菱ケミカル, JSR(QPARCの検討の一環として)
検討の背景
光化学反応とは、人工光合成材料やOLED材料の設計等に関わる産業的に重要な反応です。古典コンピュータは、光化学反応を予測する上で避けては通れない「励起状態」計算を苦手としており、今後の精度の向上は難しいと考えられてきました。これに対し、量子コンピュータを用いれば、さほど大きなコストをかけることなく励起状態計算が可能です。
実施内容
現在の量子コンピュータは産業的に重要な分子をまだ扱えないため、関連する小さな「系」を設定し、その「系」で実際に正しく計算できるかを検証しました。その結果、現在提案されているアルゴリズムを用いれば化学精度で計算可能とわかり、今後量子コンピュータの性能発展に伴い、 より大きな「系」に適用可能という見通しを立てることができました。

USE CASES / 02
周期系計算への応用検討
CLIENT
三菱ケミカル、豊田中央研究所、住友金属鉱山(QPARCの検討の一環として)
検討の背景
周期系のエネルギー計算は、半導体やエネルギー変換材料など、固体機能材料の設計にとって、なくてはならない計算手法です。産業的なニーズは高く、これまでも数多くの手法が提案され、材料設計の現場で実際に活用されていますが、重金属を含む系等、古典コンピュータでは太刀打ちできない系もまだ数多く残されています。
光化学反応と同じく、量子コンピュータを用いれば古典コンピュータが苦手とする系を扱える可能性がありますが、周期系計算には数多くの量子ビットが必要という大きな課題が存在します。そのため、これまでは実応用を本格的に検証した例は僅少でした。近年、ようやく周期系に量子コンピュータを適用した例が出てきたため、本プロジェクトではこの手法をいち早く活用し、有用性検証を試みました。
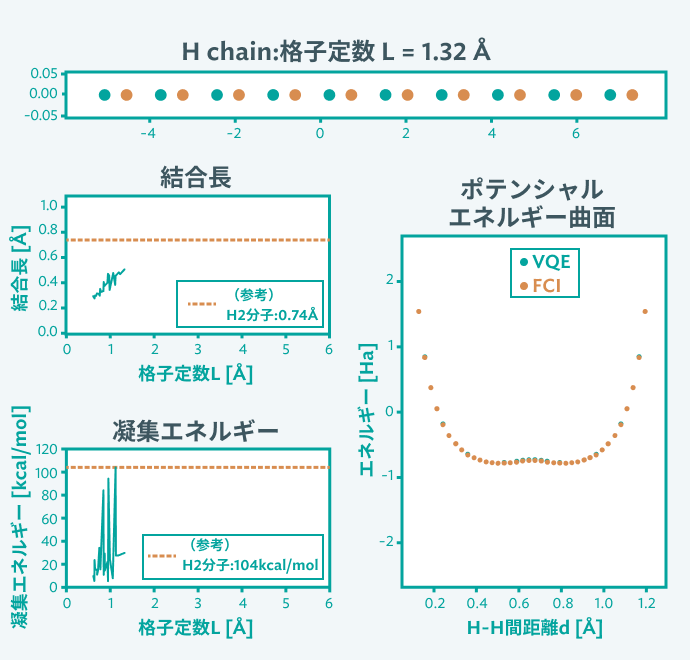
実施内容
現在の量子コンピュータの制約を考慮し、まずは水素原子2つのモデルを設定し、パイエルス転移が現れるかどうかを検証しました。その結果、水素分子から水素原子へのパイエルス転移が確認でき、現在提案されているアルゴリズムで十分な精度の計算が可能と確認できました。一方でQubit数削減は大きな課題であり、固体材料の設計を見据えると、Qubit数の削減手法の開発が不可避であることも示唆されました。今後量子コンピュータの性能発展に伴い、より大きな「系」に適用可能という見通しを立てることができました。
Partners
Qamuyのご利用について
Qamuy のご利用を検討されるお客様や下記のような疑問など、
どうぞお気軽にお問い合わせください。
- より詳しい説明を聞きたい
- 抱えている課題が解決できるか知りたい
- 導入費用について知りたい