When simulation stops scaling: Where classical modeling breaks down in complex materials

Computational chemistry plays a key role in industrial R&D, helping teams predict material properties without relying solely on time-consuming, costly, or hazardous experiments.
However, as systems grow in complexity, so do the computational challenges. Accurately modeling real-world materials — for example in catalysis, energy materials, or advanced chemicals — can quickly become prohibitively expensive, even with modern HPC resources.
In particular, certain types of systems, where electronic interactions are strongly coupled, remain difficult to treat with classical methods. In practice, this can mean that simulations fail to reproduce experimental observations or require trade-offs that limit their usefulness in real-world applications.
This webinar explores where these bottlenecks arise, how they appear in industrial R&D workflows, and how teams attempt to address them today. It also introduces how new computational approaches, including quantum computing, are being explored as potential ways to tackle these challenges.
What this webinar covered
- Where and why classical simulation methods break down as system complexity increases (e.g. strongly correlated electrons, system size scaling)
- How these limitations show up in practice: cost vs. accuracy trade-offs, failed predictions, and constrained decision-making in R&D workflows
- How teams address these challenges today, and where emerging approaches like quantum computing may fit into future simulation strategies
Who was this webinar for
- Computational chemists and materials scientists
- R&D teams using simulation and modeling
- HPC and scientific computing specialists
About the speaker

Mathias Mikkelsen
Research Scientist, QunaSys
Mathias Mikkelsen is a Research Scientist at QunaSys, where he works on quantum algorithms for chemistry simulations. He holds a master in physics and mathematics from Aarhus University and a PhD in physics from Okinawa Institute of Science and Technology. His prior research spans quantum mechanics, few-body physics, many-body physics, quantum dynamics and chaos - a common theme being experimental relevance in cold atomic gases. He has publications in Physical Review Letters, New Journal of Physics, and other leading journals. Before joining QunaSys, he held a postdoctoral research position at Kindai University.
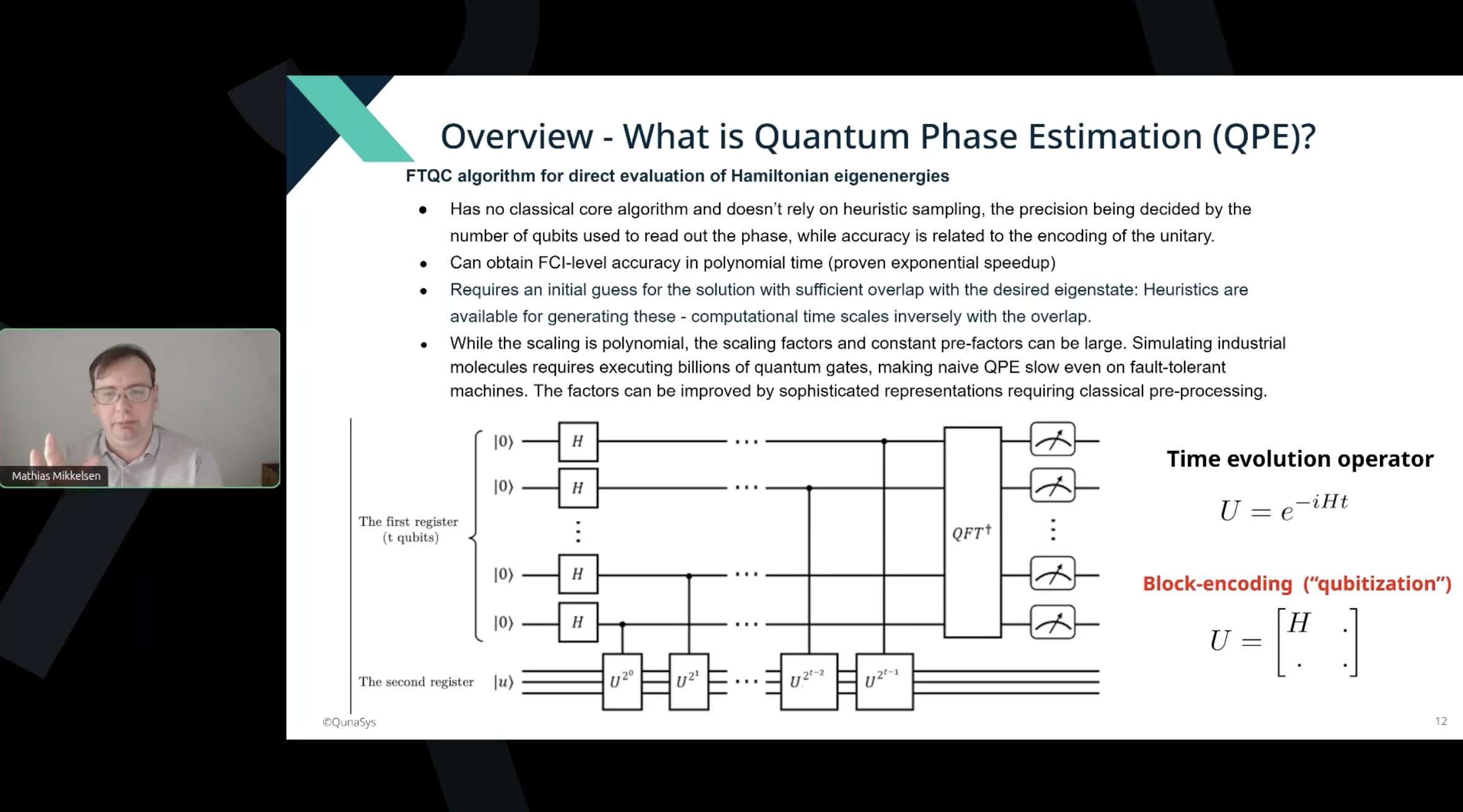
Sample slides