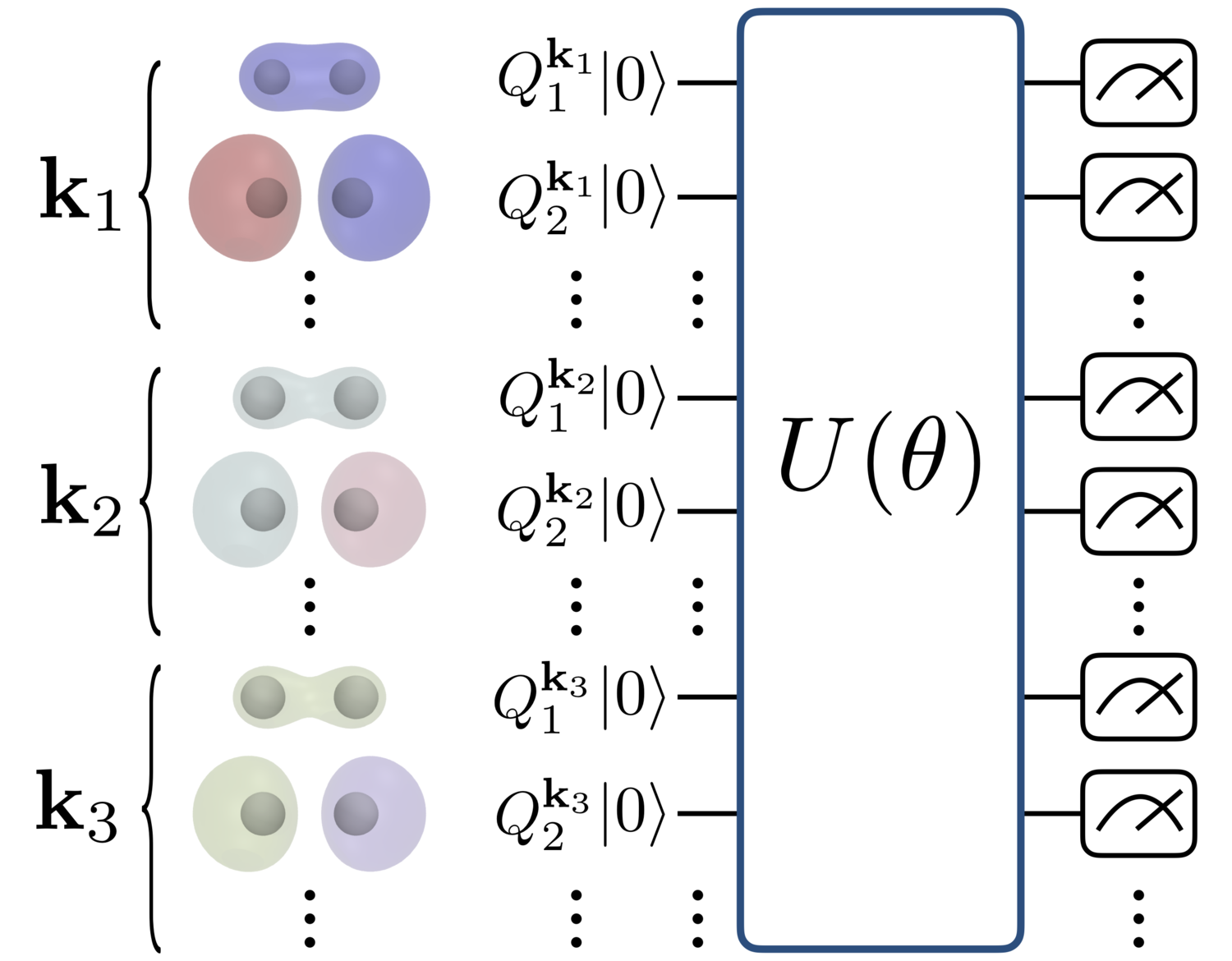
We propose a method for calculating the energy and band structure of solid-state systems using a quantum computer.
Dr. Nakagawa at QunaSys, Dr. Yoshioka at RIKEN (former QunaSys intern), Dr. Ohnishi at JSR Corp., and Dr. Mizukami at Osaka Univ (QunaSys advisor) posted a preprint on the use of variational quantum algorithms to calculate the band structure of solid-state systems.
The on-going joint research between QunaSys Inc. and JSR Corp. has led to the work.
"Variational Quantum Simulation for Periodic Materials"
https://arxiv.org/abs/2008.09492
Background
In solids, one of the three states of matter, atoms and molecules are arranged periodically, and various properties of the matter emerge depending on what they are and the way how they are arranged (i.e., crystal structure). Solid-state physics, which predicts the properties of solid material from its crystal structure, is essential for the development of new materials and has been actively researched by both academics and industry. At present, density functional theory (DFT) is widely used in theoretical calculations in solid-state physics and has achieved great success. However, it is also known that there are still important cases in which DFT has difficulties, such as the band gap of semiconductors.
The application of quantum chemical calculations, which try to solve the equation that governs a material in a more direct way compared with DFT, to solid-state physics has begun to attract attention. Quantum chemical calculations have the following advantages over DFT: (1) we can perform first-principle calculations without any empirical parameter, and (2) we can systematically improve the calculation accuracy by sophisticating the method in quantum chemical calculations.
Problems
However, the application of quantum chemical calculations to solid materials is not so easy because the amount of computational resources required to perform it gets large even for simple systems. Here the idea of using the Noisy Intermediate-Scale Quantum (NISQ) device, or a primitive quantum computer which has been rapidly developed in recent years, to quantum chemical calculations for solid materials comes out. The application of NISQ devices has been actively explored for molecular systems, but no research so far focuses on its application to solid materials regardless of its industrial importance.
Methods
In our preprint, we apply one of the most promising algorithms for NISQ devices, namely, variational quantum eigensolver (VQE), to calculating the energy and band structure of a model system of solid materials. The model system is composed of hydrogen atoms that periodically line up in one or two dimensions. We adopt an extension of the trial wave function called UCC-SD (unitary coupled-cluster singles and doubles) to solid materials and numerically simulate the output of a quantum circuit so that we can estimate what energy values could be obtained when the VQE is actually run on a quantum computer.
For numerical calculations, we used PySCF (https://github.com/pyscf/pyscf), an open-source library of chemical calculations, and Qulacs (http://docs.qulacs.org/en/latest/), a fast classical simulator of quantum circuits maintained by QunaSys.
Results
Our numerical simulations show that the ground-state (most stable) energies of the model system obtained by the VQE agree well with the exact values. Furthermore, by changing the distance between the hydrogen atoms, the VQE method was found to give energies close to exact ones even in a region where the CCSD method, a typical method of quantum chemical calculations using classical computers, returns wrong energies. In addition to the ground state energies, we also applied the so-called quantum subspace expansion method to calculating the (pseudo-)band structure. As a result, we confirmed that the energy gap can be estimated more accurately than the standard DFT (PBE) method.
Outlook
Our work (preprint) has provided a first step towards the use of quantum computers for quantum chemical calculations of solid materials. Since the application of quantum computers to solid-state materials can have a huge impact on a broad range of industrial fields, our work will trigger further exploration and analysis of the possibility of the application.